Computational Studies The usage of computational methods has become very important since the last two decades of the 20th century. In the event of lack of experimental data, computational methods offer a very reliable solutions. Even when experimental data are available, computational methods have become complementary tools for validation of the results. In other words, both experimental techniques and computational tools are complementary to each other. In our research laboratory, the computational methods are used to compute the kinetic parameters for the reactions of OH radicals, Cl atoms and NO3 radicals with varieties of compounds of interest in the Earth's atmosphere and combustion. Experimentally one can measure the overall rate coefficient for a reaction. But the information on the contribution of each site in the compound cannot be drawn so easily. Measurements with isotopic substituents should be carried out and they are very expensive. The site-specific contribution to a reaction can be easily estimated using computational methods. In addition, the contribution of type of reaction viz. addition or abstraction to the global rate can be estimated using computational methods. The transition state theory, variational transition state theory with necessary tunneling corrections are used for the calculation of rate coefficients. The branching ratios of varieties of channels in a reaction are estimated over the temperature using the computational studies. In the combustion process, understanding the mechanism is a challenging task especially in the high-temperature regime. The chemical kinetic simulations are an alternate way of understanding the decomposition mechanism of a compound. A proposed set of reactions with the known kinetic parameters can be used to compute the concentration of each and every species involved in the reaction. Kinetic situations can give us a guideline on the mechanism of a reaction. These studies are carried out in our laboratory to support the data obtained in the experimental facilities.

Book Now
Contact Info
- Department of Chemistry, Indian Institute of Technology Madras, Chennai - 600036, Tamil Nadu, India.
- +91 44 2257 4231
- rajakumar@iitm.ac.in
THE RK GROUP
Research Interests
Computational Chemistry
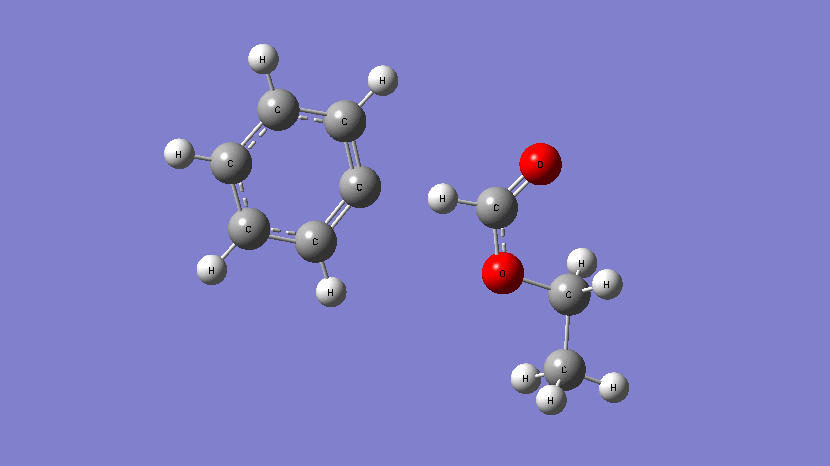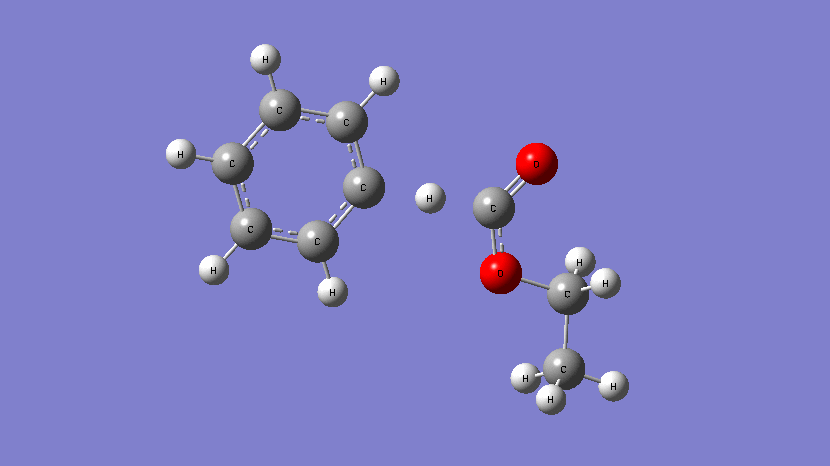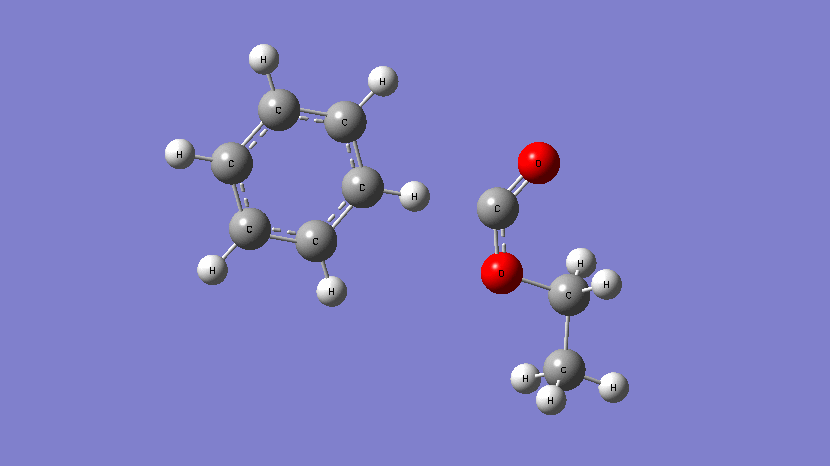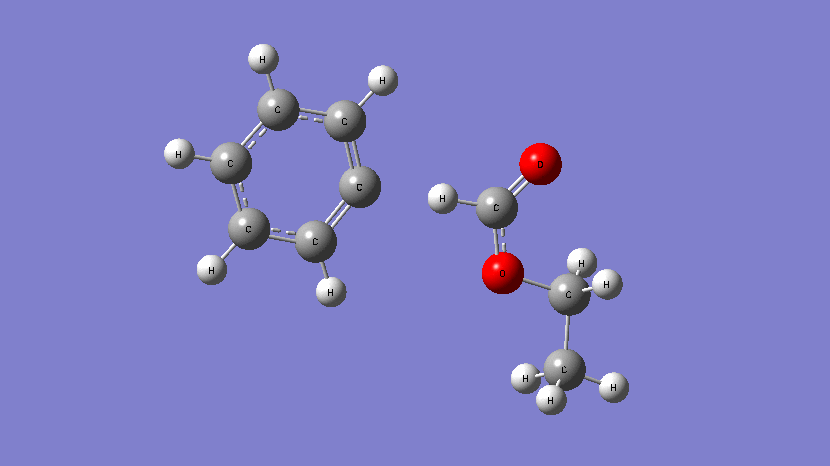

This movie shows the transition state obtained during the addition of Cl atom at a terminal position of 2,2,2-trifluoroethyl methacrylate. (Kumar et al., J. Phys. Chem. A. 2019, 123, 10868-10884)
Pressure dependent plots for the various reactions of R1OO• radicals at temperatures of 400, 1000 and 1500 K calculated using Rice-Ramsperger-Kassel-Marcus (RRKM) method which is incorporated in the MESMER 5.2 code (MESMER: Master Equation Solver for Multipotential Energy Well Reactions). [Kuzhanthaivelan et al., Combustion and Flame 2020, 219, 147-160]
To perform the computational calculations, at present we are using the AQUA supercluster, which is an indigenous facility provided by High Performance Computing Environment (HPCE), PG Senapathy Centre for Computing Resources at IIT Madras. The work by Prof. Rajakumar and his group was recently published in the HPCE highlight. HPCE Highlight of Prof. Rajakumar and his group